

Симптомы
Симптомы синдрома Ретта возникают не сразу, после рождения дети выглядят здоровыми и развиваются согласно возрасту
Заподозрить наличие патологии очень сложно, единственное, на что можно обратить внимание:
- Незначительная мышечная гипотония
- Потливость ладоней
- Незначительное снижение температуры тела
- Бледность кожи.
После 4-5 месяцев возникают первые признаки болезни – ребенок не переворачивается, не пытается ползать или сидеть.
У некоторых детей признаки заболевания возникают после 1,5-2 лет, они теряют уже освоенные навыки, перестают разговаривать, ходить, понимать обращенную речь и так далее. Но если в этом возрасте развитие ребенка, хотя и очень замедленное, все же возможно, то после 4-х лет оно полностью прекращается.
Постепенно у больного возникают все характерные признаки синдрома Ретта:
- Умственная отсталость – ребенок полностью теряет способность к обучению, если до болезни у него были освоены определенные навыки, то по мере развития заболевания они постепенно утрачиваются. Больные по умственному развитию напоминают детей первого года жизни, с сохранением только основных функций. При этом они сохраняют свою эмоциональность, остаются привязанными к членам своим семьи, узнают окружающих, радуются им.
- Гипотония – мышечная слабость мешает детям нормально ходить, сидеть, выполнять какие-либо движения. У них нарушается координация движений, исчезает целенаправленная деятельность.
- Сколиоз – ослабление мышц вызывает искривление позвоночника.
- Специфические движения рук – самый характерный признак синдрома Ретта. У детей появляются монотонные «перебирающие» движения, кажется, что он «моет руки» или трет что-то.
- Судороги – у детей часто возникают эпилептические припадки с кратковременной потерей сознания или без.
- Микроцефалия – из-за остановки роста и развития головного мозга и костей черепа у детей голова не увеличивается в размерах и остается непропорционально маленькой.
- Замедление и остановка роста частей тела – также замедляется общий рост и развитие организма.
- Нарушение работы внутренних органов – характерно возникновение проблем с пищеварением, дыханием, сердечно-сосудистой системой.
Если диагноз синдрома Ретта не был выставлен вовремя, то с течением времени установить его становится сложнее, заболевание могут спутать с умственной отсталостью, аутизмом, другими психическими патологиями или нарушениями двигательной сферы.
Без обследования внутренних органов и использования других дополнительных методов исследования легко спутать синдром Ретта и аутизм. Для обоих этих заболеваний характерны утрата приобретенных навыков, отсутствие зрительного контакта, потеря речи, беспричинные крики и плач и так далее.
Стадии
Условно течение болезни разделяют на 4 стадии:
- От 4-х мес до 2 лет – наблюдается отставание в росте и развитие ребенка, замедление роста головы и конечностей, гипотония, снижение познавательных способностей.
- 2-3 года – утрата приобретенных навыков, беспричинные крики, появление стереотипных движений, возможно возникновение судорог.
- 3-10 лет – выраженное отставание в умственном развитии, судорожные припадки, нарушение координации движений, моторики, возникновение сколиоза.
- После 10 лет – утрата способности к самостоятельному передвижению, нарушение работы внутренних органов, потеря массы тела.
Кроме классической формы заболевания выделяют несколько атипичных форм:
- Мозаичная форма синдрома Ретта – при этой форме у детей наблюдаются неврологические признаки заболевания, но нет выраженной мышечной слабости. Больные сохраняют способность самостоятельно двигаться, но у них наблюдается нарушение мелкой моторики.
- Синдром с сохраненной речью – эту форму отличает сохранение речи и умственных способностей.
- Врожденный – до сих пор не доказано, точно ли это «чистый» синдром Ретта или его сочетание с другими неврологическими заболеваниями.
- Синдром Ретта у мальчиков – раньше считалось, что синдром Ретта встречается только у девочек, но проведенные исследования доказали, что он может развиться у мальчиков с «лишней» Х-хромосомой. Такая аномалия наблюдается при синдроме Клайнфельтера.
Анализ крови и определение группы крови с резус-фактором
Подобные исследования дают представление об общем состоянии здоровья будущей мамы. Они позволяют избежать анемии и устранить возможные воспаления
Также очень важно определить группу крови и резус-фактор у обоих родителей. Это позволит устранить возможный серологический конфликт
Так происходит в тот момент, когда ребенок наследует резус — фактор от отца, а клетки крови матери лишены его. Тогда иммунная система женщины рассматривает эритроциты ребенка как враждебные. Своевременная диагностика проблемы помогает устранить риски для малыша, возникающие в результате серологического конфликта. Поэтому определение резус — фактора у обоих родителей очень важно для врача еще до беременности или на ранней ее стадии.
Синдром Ретта — лечение
Несмотря на интенсивность специальных исследований, на данный момент не существует специального лечения синдрома Ретта, воздействие, таким образом, сосредоточено на симптоматическом лечении. В связи с ухудшением физической инвалидности (сколиоз — наиболее распространенное осложнение этого состояния) у девочек проводится реабилитация и физиотерапия.
Родителям детей с синдромом Ретта поручают контролировать положение тела ребенка и сохранять правильное положение во время повседневной деятельности, что очень важно как для профилактики сколиоза, так и для контроля его прогрессирования. Чем больше боковое искривление позвоночника, тем больше сложности в областях поддержания равновесия и вертикального положения, общей подвижности и функционирования внутренних органов. . Девочки с синдромом Ретта испытывают огромный барьер и трудности в общении — они редко используют слова, и хотя после прекращения аутистического поведения они открываются миру и проявляют коммуникативные намерения и интерес к другим людям, постепенный нейромоторный регресс, повышение мышечного тонуса и трудности в выражении чего-либо с помощью жестов и слов, мешают им нормально контактировать с окружающей средой
Поэтому важно развивать альтернативную и вспомогательную коммуникацию.
Девочки с синдромом Ретта испытывают огромный барьер и трудности в общении — они редко используют слова, и хотя после прекращения аутистического поведения они открываются миру и проявляют коммуникативные намерения и интерес к другим людям, постепенный нейромоторный регресс, повышение мышечного тонуса и трудности в выражении чего-либо с помощью жестов и слов, мешают им нормально контактировать с окружающей средой
Поэтому важно развивать альтернативную и вспомогательную коммуникацию. . Благодаря терапевтическим, ревалидационным и реабилитационным воздействиям, адекватным возможностям ребенка в образовании, а также части пациенток получают возможность самостоятельно функционировать
Психологи и терапевты могут быть сосредоточены на поддержке психического и социального, интеллектуального и эмоционального развития. Логопед сосредотачивается на работе по эффективному общению с окружающей средой
Благодаря терапевтическим, ревалидационным и реабилитационным воздействиям, адекватным возможностям ребенка в образовании, а также части пациенток получают возможность самостоятельно функционировать. Психологи и терапевты могут быть сосредоточены на поддержке психического и социального, интеллектуального и эмоционального развития. Логопед сосредотачивается на работе по эффективному общению с окружающей средой.










Как помочь больному
Синдром Ретта относится к числу неизлечимых заболеваний, склонных к прогрессированию. Продолжительность жизни при этой патологии разная. Многие больные умирают в детстве и юности от прогрессирующей дистрофии, во время эпилептических припадков, а также из-за нарушения вентиляции лёгких, спровоцированной сколиозом. Но при должном уходе и правильном симптоматическом лечении срок жизни можно существенно продлить. Известны случаи, когда пациенты доживали до 40 и даже до 50 лет.
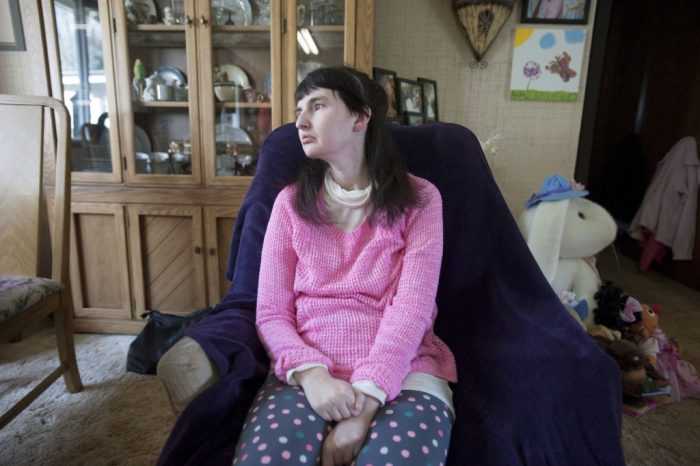
Многие больные живут 40 лет и больше
Моя врачебная практика показывает, что течение болезни может варьироваться от очень тяжёлого до относительно лёгкого. Есть больные, которые с детства почти полностью обездвижены и прикованы к инвалидным коляскам. Но другая часть пациентов долгое время сохраняет двигательные функции
И что самое важное: в их состоянии и поведении нередко наблюдается некоторая положительная динамика
Способы реабилитации
Лечение синдрома Ретта симптоматическое. Его цель — достижение ремиссии и стабилизация состояния больного. Если течение болезни относительно лёгкое, применяется амбулаторная терапия. При тяжёлом состоянии пациента госпитализируют. Основные лечебные методы:
-
Физкультура и трудотерапия — позволяют как можно дольше сохранять двигательные навыки и поддерживать мышечный тонус.
- Музыкальная терапия — оказывает успокаивающее действие, в какой-то степени компенсирует отсутствие общения с окружающими миром.
- Диета с повышенным содержанием жиров — является эффективной при дистрофии, даёт возможность больному набрать вес.
-
Массаж — применяется для предупреждения развития сколиоза и других тяжёлых ортопедических осложнений.
- Медикаментозная терапия — при приступах эпилепсии применяются противосудорожные средства, а при бессоннице — мелатонин.
-
Семейная терапия — предполагает общение родственников больного с психологом с целью стабилизации психологического климата в семье.
Как развивается заболевание
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов
Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков
Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.









Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
Причины возникновения
Точные причины возникновения синдрома Ретта до сих пор остаются предметом дискуссий, поэтому можно привести в пример несколько наиболее распространенных теорий. Все они сейчас проверяются путем проведения различных исследований, и хорошо еще, что медики хотя бы установили отличие от слабоумия и теперь движутся в правильном направлении.
Чаще всего специалисты указывают на генное происхождение болезни: они говорят, что недуг вызван мутацией генов. Данное отклонение развивается до масштабов патологии еще на этапе вынашивания плода, поэтому последствия видны уже на ранних этапах жизни.

Указывается, что повышенное количество кровных связей в родословной наверняка влияет на вероятность развития синдрома Ретта, причем процент таких связей высок лишь по сравнению с нормой – 2,5% вместо 0,5%. В пользу такого фактора, как причины возникновения патологии, говорят и ярко выраженные очаги заболеваемости – обычно это небольшие, достаточно изолированные от мира деревушки, где половые связи между членами одной семьи в прошлом вполне вероятны.
Другая группа исследователей указывает на нарушения в Х-хромосоме – ломкость одного из участков ее короткого плеча.
Доказано, что иногда такая аномалия действительно наблюдается, но ученые пока не смогли обнаружить точный участок, отвечающий за развитие патологии, равно как и установить определенную закономерность, позволяющую утверждать стопроцентную взаимосвязь причины и следствия.
Существует и третья (менее известная) теория, согласно которой синдром Ретта может быть следствием метаболических аномалий – нарушенного обмена веществ, вызванного митохондриальной дисфункцией.
Отмечается, что у всех больных данным заболеванием отмечено повышенное содержание в крови двух кислот – пировиноградной и молочной, очевидны также патологии лимфоцитов и миоцитов. Впрочем, это пока только наблюдение, и специалисты сейчас не могут сказать, является ли это причиной возникновения синдрома Ретта или его следствием.
Прогноз при синдроме Ретта

Младенцы мужского пола с патогенными MECP2 мутациями, как правило, умирают в течение первых 2 лет от тяжелой энцефалопатии, если в их генетическом составе была найдена дополнительная X-хромосома. Такое состояние часто описывается как синдром Клайнфельтера, или соматический мозаицизм.
Девочки способны прожить дольше в силу наличия дополнительной Х-хромосомы не подвергнутой мутационным изменениями. Без нормального гена, который способен обеспечить нормальные белки в дополнение к аномальным, что вызвано мутацией MECP2, XY-кариотип мужского пола не может замедлить развитие болезни. По этой причине, неспособность многих детей мужского пола с мутацией MECP2 выжить в перспективе проявляется наиболее ярко.
Взрослые женщины могут жить до 40 лет или более. Лабораторные исследования при синдроме Ретта у них могут показывать следующие аномалии:
- ЭЭГ нарушения от 2 летнего возраста, в процессе всей жизни;
- наличие атипичных гликолипидов в мозге;
- повышенные уровни бета-эндорфинов и глутамата;
- снижение субстанции Р;
- снижение уровня нервных факторов роста.
Доля смертности достаточно высока, но большинство летальных исходов не имеют определенных причин. В некоторых случаях смерть является результатом:
- спонтанной дисфункции ствола мозга;
- остановки сердца, вероятно, из-за длительного интервала QT-синдрома, желудочковой тахикардии или других типов аритмий;
- судорог;
- перфорации желудка.
Диагностика

Синдром Ретта — это клинический диагноз, то есть для его выставления необходим ряд клинических признаков. Международная ассоциация по изучению синдрома Ретта предложила диагностические критерии данного заболевания. Среди них выделено несколько групп:
- необходимые (их наличие обязательно для установления диагноза);
- дополнительные (их наличие подтверждает диагноз, но не является обязательным);
- исключающие (наличие хотя бы одного из них отвергает синдром Ретта).
К необходимым критериям относят:
- отсутствие патологии у плода, выявленной во время беременности и в первые 7 дней жизни, нормальный период родов;
- нормальная окружность головы при рождении и замедление темпов ее прироста между 5 месяцами и 4 годами;
- утрата целенаправленных движений руками в возрасте от полугода до 30 месяцев, совпадающая по времени с утратой навыков общения;
- появление стереотипных движений рук после утраты целенаправленных;
- нарушение речи и задержка умственного развития;
- нарушения ходьбы, выявляющиеся в возрасте 1-4 лет.
Не всегда при синдроме Ретта присутствуют все необходимые критерии заболевания. Такие формы болезни относят к неполным, обычно они характеризуются более легким течением.









К дополнительным критериям относят:
- наличие расстройств дыхания в виде периодов учащенного дыхания, сменяющихся остановками. Появление расстройств дыхания только в период бодрствования;
- эпиприпадки;
- нарушение тонуса мышц в сочетании с их атрофией;
- нарушение кровотока в периферических отделах конечностей;
- сколиоз;
- задержка роста;
- маленькие недоразвитые ступни (вследствие нарушения ходьбы стопа не достигает нужной степени развития);
- наличие изменений на электроэнцефалограмме в виде эпиактивности даже при отсутствии явных эпиприпадков.
Исключающие критерии:
- внутриутробная задержка роста;
- увеличение размеров внутренних органов (органомегалия);
- ретинопатия, или атрофия дисков зрительных нервов;
- малые размеры головы (микроцефалия) уже при рождении;
- подтвержденное перинатальное повреждение головного мозга (например, травма во время родов);
- перенесенная тяжелая инфекция или черепно-мозговая травма, в связи с которыми появились неврологические расстройства;
- существование иного (не синдрома Ретта) неврологического или метаболического подтвержденного заболевания, имеющего прогрессирующий характер.
Критерии диагностики
Диагноз синдрома Ретта основывается на распознавании характерной клинической картины. Для этого Международной ассоциацией по изучению синдрома Ретта предложена группа диагностических критериев, которые разделены на необходимые, дополнительные и исключающие. Классическая форма синдрома Ретта может быть диагностирована, если у пациента присутствуют все необходимые критерии. Следует отметить, что женский пол не входит в их число, поскольку это могло бы уклонить врачей от поиска мальчиков с синдромом Ретта. Вторая группа состоит из дополнительных критериев, многие из которых обычно имеются у больных, но ни один из них не является обязательным для постановки диагноза. Третья группа — исключающие критерии, одного из которых достаточно, чтобы отвергнуть синдром Ретта у пробанда.
Диагностические критерии синдрома Ретта (по Trevathan et al., 1998) включают необходимые критерии, среди которых нормальные пренатальный и перинатальный периоды, нормальная окружность головы при рождении с последующим замедлением роста головы между 5 месяцами и 4 годами; потеря приобретенных целенаправленных движений рук в возрасте от 6 до 30 месяцев, связанная по времени с нарушением общения; глубокое повреждение экспрессивной и импрессивной речи и грубая задержка психомоторного развития; стереотипные движения рук, напоминающие выжимание, стискивание, хлопки, “мытье рук”, потирание, появляющиеся после потери целенаправленных движений рук; нарушения походки (апраксии и атаксии), выявляющиеся в возрасте 1 — 4 лет. Диагноз считается предварительным до двух-пятилетнего взраста.
Хотя исследователи единодушны в том, что в развитии патологии наследственные факторы играют существенную роль, их мнения относительно механизмов наследования синдрома Ретта расходятся
Дополнительные критерии включают дыхательные расстройства в виде периодических апноэ во время бодрствования, перемежающихся гипервентиляцией, форсированного изгнания воздуха и слюны, аэрофагии; судорожные припадки; спастичность, часто сочетающуюся с дистонией и атрофией мышц; периферические вазомоторные расстройства, сколиоз, задержку роста, гипотрофичные маленькие ступни, электроэнцефалографические аномалии (медленный фоновый ритм в состоянии бодрствования и периодическое замедление ритма (3-5 Гц), эпилептиформные разряды без или с наличием клинических судорог).
Наконец, к исключающим критериям относят очевидность внутриутробной задержки роста, органомегалию или другие признаки болезней накопления, ретинопатию или атрофию дисков зрительных нервов, микроцефалию при рождении, доказательство перинатально приобретенного повреждения мозга, существование идентифицированного метаболического или другого прогрессирующего неврологического заболевания и приобретенные в результате тяжелой инфекции или черепно-мозговой травмы неврологические нарушения.
У ряда больных клинические признаки не соответствуют полностью классическому течению синдрома Ретта. Эти случаи классифицируют либо как неполные, либо как атипичные формы заболевания. При неполной форме у больного присутствуют многие, но не все из необходимых симптомов. Этим характеризуются легкие варианты болезни. Атипичные формы — это случаи синдрома Ретта, которые соответствуют всем необходимым критериям диагностики, но имеют отклонения от типичного течения. В частности, при атипичной форме синдрома с ранним началом судорог эпиприпадки являются дебютом заболевания. Раннее начало эпилепсии не оказывает существенного влияния на течение и прогноз болезни, однако вызывает дифференциально-диагностические трудности. При атипичном варианте синдрома с частично сохраненной речью больные имеют некоторые речевые навыки, течение заболевания у них более мягкое, чем при классической форме, а уровень общения значительно выше. Известны также атипичные варианты синдрома с аномальным развитием ребенка с рождения, поздним началом фазы регресса, сюда же относят случаи синдрома Ретта у мальчиков.
Проведенные в отделе врожденных и наследственных заболеваний МНИИ педиатрии и детской хирургии исследования продемонстрировали значительный клинический полиморфизм синдрома Ретта. Среди 40 наблюдавшихся пациентов в возрасте от 20 месяцев до 16 лет выявлено 27 случаев классического синдрома Ретта и 13 атипичных случаев. Генеалогический анализ показал накопление в родословных пробандов случаев умственной отсталости, судорожных состояний, психических заболеваний. Эти наблюдения, по-видимому, могли являться случаями синдрома Ретта с низкой экспрессивностью.